
Bioprocessing Risks: Why Inspection Readiness Matters
March 27, 2026

Inspection‑Ready Bioprocessing: The Hidden Risk Separating Asia’s Winners from Its Costliest GMP Failures
Walk through any new bioprocessing facility in Asia, and you will see impressive cleanrooms, gleaming skids, and state‑of‑the‑art utilities. Yet the most expensive failures in life sciences rarely begin when a batch goes out of specification or an inspector issues a citation. They begin months earlier, when leadership approves a design based solely on speed and capital efficiency, assuming that compliance can be bolted on later. This hidden design debt—misaligned zoning, unresolved containment logic, unstable utilities, thin traceability, and fragile validation architecture—remains invisible until inspectors force the entire asset to operate as a single, defendable system. By then, the cost of correction is no longer technical; it is strategic.
Why does this matter now? Because Asia’s bioprocessing boom is entering its most competitive phase. Markets across South‑East Asia are adding capacity at an unprecedented pace, driven by demand for biologics, vaccines, and advanced therapies. However, capacity is no longer the differentiator. A finished building is not a defensible asset; an approvable facility is. Facilities that cannot explain their design logic under EU‑GMP, US FDA, or PIC/S scrutiny become slower to approve, harder to partner with, and exponentially costlier to stabilize. The result: investors lose patience, commercial timelines slip, and the firm’s reputation suffers.
That is why the industry is closely watching Taiwan. As highlighted in a recent BioPharma APAC analysis, Taiwan’s next phase of bioprocessing growth is not just a story of scale; it is a masterclass in regulatory architecture. Rather than treating compliance as a late-stage hurdle, Taiwan is building export-ready manufacturing ecosystems anchored entirely on the credibility of early-stage inspection readiness.
The Hidden Cost of "Design Debt"
Picture a project team racing toward mechanical completion. To stay on schedule or to save the initial CapEx, they compress HVAC design reviews, accept “good‑enough” zoning, and defer data‑integrity architecture, thus falling into the “Design Debt” trap. On paper, the facility meets its milestones; in practice, it has baked in latent exposure. When validation begins, the fragility shows. Experts must be brought in to redesign flows and requalify systems.
What begins as a minor HVAC configuration choice or a personnel flow shortcut inevitably becomes a recurring operating inefficiency. What is deferred in contamination control or digital traceability later reappears as severe inspection pressure and a loss of execution confidence. Growth often conceals this debt until the cost of reversal requires tearing down walls or replacing entire utility systems. Not to mention, the cost of bringing in outside consultants to remediate quality issues is high for companies of any size, and reactive remediation is several times more expensive than proactive quality management. The remedial effort impacts daily operations, diverts resources, and erodes morale.
Financially, design debt manifests as direct and indirect quality costs. A McKinsey‑sourced analysis of the medical‑device sector shows that quality costs fall into three categories — prevention, appraisal, and failure—and together they consume 6.8–9.4% of annual sales, amounting to US$ 30–43 billion. Routine internal failures—scrap, rework, and deviations—account for 2.1% of sales, while remediation costs for investigations and field actions add 0.4–0.7%. Non‑routine failures, such as major recalls and consent decrees, consume 1.9–2.5% of sales, roughly US$17 billion in 2022. Indirect costs tied to plant shutdowns and market‑cap erosion can reach US$ 1–3 billion. Clearly, skimping on early risk review is a false economy.
Beyond numbers, the reputational fallout can be devastating. Pharmaceutical Technology notes that firms receiving warning letters face continuous regulatory scrutiny that can take two to three years to resolve. Financial institutions may raise interest rates; suppliers may demand advance payments; top talent may leave.
“Most GMP failures are not discovered. They are designed.”
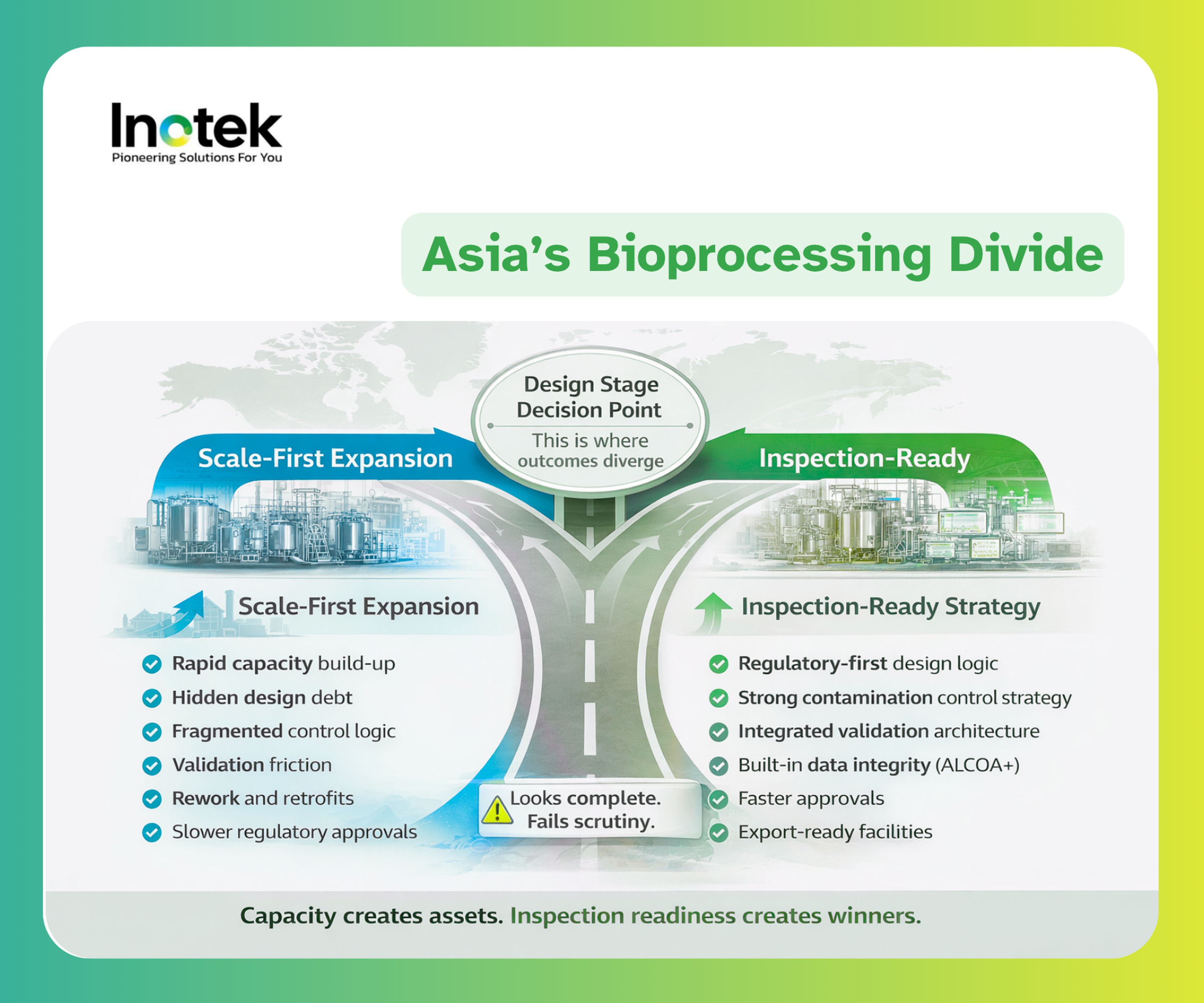
From Scale to Inspection Readiness: Asia’s Emerging Divide
Early investors in Asia’s bioprocessing surge assumed that more square meters and more bioreactors would guarantee market access. Today, that assumption is being challenged. Installed capacity without an inspection‑ready design is a stranded asset. The fastest way to lose investor confidence is to build a facility that looks complete yet crumbles when regulators ask, “Show me the scientific justification for this flow.”
Common Design Traps
Over‑generalized cleanroom philosophy:
Viral vectors, mRNA, and cell therapies have different contamination profiles. Applying a single segregation model across all modalities either inflates CapEx through unnecessary barriers or leaves process risks inadequately controlled. The result is an expensive facility that cannot handle its intended products.
Misaligned personnel and material flows:
Early in design, flows often follow the shortest path rather than the path that minimizes cross‑contamination. Inspectors immediately question flows that intersect with return ducts or cross high‑risk areas without buffer zones. Redesigning flows post‑construction is disruptive and costly.
Unresolved containment strategy:
Decisions about HVAC, pressure cascades, and utilities are often deferred to a later date. Under‑design leads to unstable utilities; over‑design creates operating inefficiencies. A robust Contamination Control Strategy (CCS) is a capital decision, not just a quality document.
Fragmented digital architecture:
Many teams assume that data integrity can be bolted on through SOPs. In reality, ALCOA+ principles demand that audit trails, exception handling, and electronic signatures be architected into automation and historian systems from day one. Weak data governance surfaces as batch‑release friction and inspection vulnerability.
What Taiwan Gets Right: Engineering Regulatory Credibility
Taiwan is engineering the conditions that preserve asset credibility under intense regulatory scrutiny. In a sector where minor design misjudgments can surface as approval drag, their ecosystem model is highly instructive for any firm looking to build in Asia.
1. Regulatory Convergence as a Financial Strategy: The true cost of weak regulatory convergence is the silent premium paid throughout the asset lifecycle when design decisions fail to travel well across global jurisdictions. If a facility is built only to local standards, it becomes a "stranded asset" when the company decides to export to the EU or the US. Taiwan reduces this premium by operating closer to globally legible GMP expectations (PIC/S, EU-GMP, USFDA) from the outset. This "inspect-once, export-anywhere" logic is a powerful financial hedge.
2. Anticipating Convergence in Facility Design: When international expectations are anticipated early, facilities carry less latent exposure in cleanroom philosophy, aseptic interventions, and validation architecture. This is where regulatory-driven design matters most: before technical decisions harden into permanent, expensive compliance debt.
3. Policy-Led GMP Infrastructure: Taiwan’s approach ensures clusters are built with proximity to validation capabilities and quality support. They recognize that a facility is only as good as the ecosystem that supports its qualification. This reduces the risk of fragmented handoffs—where the builder finishes, the validator starts, and neither understands the other’s assumptions.
Why Some Facilities Will Scale but Fail Inspections
Many facilities that look robust on paper harbor unresolved assumptions. Design‑led GMP gaps—such as poor zoning, flawed flow paths, or impractical layouts—lock in risks that are expensive to reverse. Weak validation strategy architecture arises when URS, design, and qualification documents do not align, turning validation into a patchwork exercise. Inadequate QMS integration forces investigations and CAPA to compensate for design mistakes, leading to years of regulatory scrutiny. Finally, talent and execution constraints—teams with technical skills but lacking inspection‑level judgement—allow blind spots to persist. These factors explain why some facilities will scale quickly but falter when inspectors demand coherence.
Regulatory expectations now require facilities to justify how operational robustness was anticipated at the concept stage. Inspectors are no longer looking for "how you fix problems"; they are looking for "how you engineered the facility so the problem couldn't happen in the first place." If you are relying on procedural workarounds to fix a design flaw, you have already lost the inspection.
Advanced Modalities Are Repricing Facility Risk
The shift toward advanced modalities—mRNA, Cell & Gene Therapy (CGT), and next-generation biologics—reprices risk across the entire asset. Conventional GMP logic, which worked for simple oral solids or stable liquids, is insufficient for these structurally complex platforms.
- Viral Vector vs. mRNA Segregation: These platforms have vastly different contamination profiles. Overgeneralized cleanroom logic either creates unnecessary CapEx through over-engineering or leaves process risks insufficiently controlled, leading to catastrophic batch losses.
- Closed vs. Open Processing: There is a dangerous myth that "closed systems" solve all contamination issues. In reality, closed processing only works when system boundaries and transfer integrity are architected correctly. If the facility layout doesn't support maintaining these boundaries, the "closed" system becomes a false sense of security.
- Contamination Control Strategy (CCS) as a Capital Decision: Under Annex 1, a CCS is not a quality document—it is a capital-protection decision. Once your barriers, pressure cascades, and utility logic are locked, you have priced in either total control or recurring exposure.
“By the time validation struggles, the real mistake is already built.”
Where Asset Resilience is Won (or Lost)- Long Before Inspection
Asset resilience is determined months or years before an inspector walks through the door. It is built upon three critical pillars that are often overlooked in the early "construction" phase.
1. Cross-Regulator Acceptance (EU, USFDA, PIC/S): What gets missed is the cost of regulatory reinterpretation. If your facility logic and validation architecture do not translate cleanly across major regulatory expectations, you are forced into a cycle of "patchwork compliance." Every time you enter a new market, you have to reinterpret your data and your design. This slows approvals, weakens partner confidence, and forces leadership to engage in late-stage defensive spending. Resilience means building a design that is "region-agnostic."
2. Data Integrity and Digital Traceability (ALCOA+): Data integrity is an architectural issue, not a downstream quality topic. You cannot "fix" data integrity with an SOP if the underlying system architecture is fragmented. Weak audit-trail strategy, poor exception handling, and fragmented historian logic surface as friction in batch releases and inspection vulnerabilities. If your digital traceability is not engineered into the facility’s automation layer from Day 1, you are building an asset that will struggle to maintain self-governance.
3. Talent Depth and Inspection Experience: The most underestimated exposure is the quality of decisions made before the first brick is laid. Teams without sufficient real-world inspection exposure tend to normalize weak assumptions. They approve design logic that "seems fine" but is actually impossible to defend during a high-stakes audit. This is precisely why early regulatory advisory matters: not to explain requirements (which anyone can read), but to challenge blind spots before they harden into approval delays.
“Inspection pressure does not create risk. It reveals it.”
How to Know If Design Debt Is Already Embedded
Many facilities do not recognize design debt until validation or inspection exposes it. However, the warning signs are usually present much earlier.
If your project currently:
- cannot clearly explain zoning and flow logic without relying on procedural controls
- is deferring validation strategy until after detailed design
- assumes data integrity will be addressed through SOPs rather than system architecture
- requires “workarounds” to justify material or personnel movement
- relies on multiple interpretations of regulatory expectations across teams
Then design debt is likely already embedded in the asset. At this stage, the question is no longer whether risk exists but whether it can still be corrected without significant cost, delay, or redesign.
The earlier this is identified, the more options remain. The later it is discovered, the fewer choices exist.
Building Inspection-Ready Assets Before Risk Gets Locked In
Leadership teams have an opportunity to turn compliance into a competitive advantage. The following decision points, distilled from both industry experience and Inotek’s regulatory‑first practice, highlight where early interventions deliver the greatest return:
- Zoning and flow logic: Ask how materials, personnel, and waste will move, where flows intersect, and which barriers or airlocks are needed. Weak zoning is among the most common reasons for inspection failures.
- Containment and Clean‑Utility Strategy: Determine the clean‑utility philosophy early—pressure cascades, air changes per hour, clean steam, and water for injection. Align decisions with the highest product exposure and regulatory expectations.
- Validation architecture: Decide whether the project will follow a conventional commissioning–qualification–validation (CQV) model or an integrated approach. Ensure that User Requirement Specifications (URS), design qualification, and operational qualification are linked; otherwise, validation will expose misalignment rather than confirm control.
- Automation and data governance: Select manufacturing execution systems and automation platforms that support data integrity out of the box. Establish standardized audit trails, exception handling, and electronic signatures.
- Quality management system (QMS) alignment: The QMS must reflect physical reality. Paper‑thin compliance—procedures that cannot be executed in the designed space—invites deviations. Mature systems integrate CAPA, deviation management, and change control with actual operations.
- Talent and training plan: Anticipate the skills needed for advanced modalities. Recruit and train personnel well before commissioning. Consider that talent depth is part of the facility’s defensibility.
These decision points are not technical details; they are strategic choices with multi‑year commercial consequences. They determine whether a facility can absorb new products, adapt to changing regulations, and withstand inspection pressure without capital‑intensive retrofits.
What Should Leaders Prioritize for Inspection‑Ready Bioprocessing?
Leadership mispricing often occurs before design freeze, when decisions are driven by throughput and timeline rather than by risk. To avoid embedding irreversibility, leaders must ensure that every zoning, utility, and automation decision is scrutinized through a regulatory lens. Translating guidelines into engineering decisions requires expert interpretation; guidelines provide principles, not blueprints. Avoiding post‑design compliance corrections means investing in concept‑stage reviews and independent design audits. Reactive remediation costs are several times higher than proactive investments
How to Know When a Facility Looks Finished but Is Not Ready?
The most dangerous assets often look complete. They meet milestones, compile documents, and project confidence. Yet serious gaps may lurk. A rigorous design review against GMP expectations calls into question whether zoning, containment, and monitoring logic truly meet global standards. A validation readiness assessment goes beyond counting protocols to ask whether the facility’s control architecture can withstand real operations. A QMS–engineering alignment check tests whether the quality and engineering teams can coherently explain how their systems interact under deviations and changes. If they cannot, the facility may appear finished, but it is not ready.
Turning Compliance Into Competitive Advantage
Asia’s bioprocessing landscape is at a turning point. The region’s growth is no longer measured by the expanse of facilities but by inspection‑ready, export‑capable facilities. Taiwan’s rise demonstrates the power of embedding regulatory credibility into design; the broader industry evidence shows the staggering cost of ignoring it. Design debt is a high‑interest loan—what saves a few weeks today can cost millions tomorrow.
Yet a regional consulting gap persists. Though South‑East Asia is attracting serious attention from biotech investors and strategic partners, advisory depth across the region remains uneven with dependence on offshore interpretation for GMP strategy and facility logic—consultants who may understand the regulations but do not understand the local execution realities or the site's specific engineering challenges. This creates a disconnect between the “ideal” regulatory requirement and the “actual” facility logic. Addressing this gap by engaging advisors with both regulatory expertise and on‑the‑ground engineering insight is essential to making inspection‑ready design a regional standard.
Leadership should therefore ask:
- Do we truly understand our facility’s control logic, or are we relying on disconnected assumptions?
- Are we comfortable explaining every design choice under EU‑GMP and US FDA scrutiny?
- Have we engaged independent expertise to challenge our decisions before they harden into irreversible commitments?
By prioritizing regulatory‑first consulting, organizations convert compliance from a cost center into a strategic asset. The result is faster approvals, lower operating costs, greater flexibility, and stronger investor confidence.
Inotek’s Role: Regulatory‑First Consulting Without Execution Bias
In most bioprocessing projects, the highest-impact decisions are made before validation begins—often without sufficient regulatory interrogation. By the time issues surface, they are no longer design questions; they are embedded costs.
This is the gap Inotek addresses. Not as an additional layer of consulting, but as the point of intervention where risk is still reversible. Unlike EPC contractors or equipment-led advisors, Inotek operates independently of execution. This removes commercial bias from decision-making and allows early-stage assumptions to be challenged without constraint.
The objective is not to support project delivery—but to ensure that what is delivered remains defensible under inspection, scalable in operation, and resilient over time.
Inotek structures its advisory into four core offerings:
Design & Engineering Consultancy—from conceptual design to detailed engineering, ensuring zoning, flows, cleanroom engineering, and utilities are aligned with global regulatory expectations.
International Regulatory Certification Facilitation—translating EU‑GMP, US FDA, WHO, and PIC/S expectations into design requirements and supporting inspection preparedness.
Quality Management Systems Consulting—embedding quality logic into facility and process design so that documentation supports audits rather than merely satisfying procedural checklists.
Validation Consulting—developing validation strategies and master plans that confirm control rather than expose design flaws.
Because Inotek does not sell equipment or manage construction, its advice is unbiased. Senior subject‑matter experts (SMEs) lead engagements and challenge early design assumptions. This model safeguards leadership from the hidden irreversibility that creeps into projects when decisions are made by execution‑biased teams.
If your design is already approved, the real question is not whether risk exists but where it is embedded, and whether it is still reversible. The value of this intervention is time-sensitive. Once zoning, flows, utilities, and validation logic are fixed, the cost of correction escalates rapidly and in many cases, becomes structurally irreversible.
In Asia's fast-growth markets, most projects do not fail due to lack of investment or intent. They fail because critical assumptions were never challenged early enough. Inotek exists to challenge those assumptions before they become permanent.
Are your facility decisions ready to withstand global regulatory scrutiny?
Do not wait for an inspection to find out that your design is a liability. Engage Inotek to evaluate your compliance risk and protect your capital before your next design freeze.
Discover more at www.inotek.co.in/contact-us.
FAQs:
1. How can leadership tell whether a bioprocessing facility is genuinely inspection-ready or only appears complete on paper?
A facility may appear complete based on project milestones yet still carry hidden exposure in zoning logic, contamination control, traceability, validation architecture, or QMS design alignment. The real test is whether its control strategy remains coherent and defensible under qualification, inspection, and routine operating stress.
2. Which design-stage decisions create the highest downstream risk for GMP inspections in advanced bioprocessing facilities?
The highest-risk decisions usually fall under segregation philosophy, pressure-cascade logic, personnel and material flows, intervention design, utility assumptions, and URS-to-validation traceability. These choices shape whether the facility can later demonstrate control or must rely on procedural workarounds to compensate for design weakness.
3. Why do advanced modalities such as mRNA, viral vectors, and multiproduct therapy platforms make conventional GMP facility logic insufficient?
These modalities change the risk profile at a structural level by increasing sensitivity around segregation, contamination control, cleaning validation, intervention strategy, and operational flexibility. A facility designed around generic bioprocess assumptions can become either unnecessarily expensive or insufficiently controlled once the actual modality-specific risks are tested.
4. What usually causes validation programmes to become fragile even when documentation appears complete?
Validation weakens when the facility was never configured to make validation persuasive in the first place. Poor layout, weak cleanability, unresolved flows, impractical interventions, and incomplete traceability often surface later as deviations, qualification friction, and inspection questions that documentation alone cannot neutralize.
5. At what stage does an external regulatory-first advisory create the most value in a bioprocessing project?
The highest value comes before design logic hardens—during concept development, early layout decisions, and initial definition of segregation, utilities, contamination-control strategy, QMS logic, and validation intent. That is the point where hidden compliance debt can still be identified and corrected without major capex, timeline, or credibility loss.



